研究背景
1960年Jennings等第一次提出心肌缺血再灌注损伤的概念,证实再灌注会引起心肌超微结构不可逆坏死,并逐渐引起医学界的高度重视。缺血心肌恢复再灌注后,病情反而恶化,引起超微结构、功能、代谢及电生理方面发生进一步的损伤,是由于在缺血损伤的基础上再次引起的损伤,因此称为缺血再灌注损伤(ischemia
reperfusion
injury,IRI)。临床上表现为闭塞的冠状动脉再通、梗死区血液灌流重建后一段时间内,有的病例发生血压骤降、心功能不全、心律失常甚至猝死等一系列病情反而恶化的现象。因此,IRI的发生机制与防治越来越引起人们的关注,并一直试图寻找能对IRI产生确切保护作用的药物。
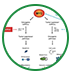
目前,缺血再灌注损伤发生的机制尚未完全阐明,研究表明自由基、钙超载、心肌纤维能量代谢障碍、中性粒细胞、血管内皮细胞、细胞黏附分子与细胞凋亡等均可能参与缺血再灌注损伤(如下图所示)。1)氧自由基(FR)生成:再灌注时产生的大量氧自由基不能被清除,它可与各种细胞成分,如膜磷脂、蛋白质、核酸等发生反应,造成细胞结构损伤和功能代谢障碍。冉擘力等从细胞水平证明早期产生的氧自由基能诱导延迟保护作用产生,其机制可能是通过早期氧化反应一方面改变SOD形态结构而提高酶的活性,诱导延迟相SOD合成增加,另一方面诱导热休克蛋白信使核糖核酸转录和持续合成,保护心肌细胞对抗细胞外氧自由基的损伤;一氧化氮合酶(NOS)产生的一氧化氮能有效对抗氧自由基的损害,延迟期心肌NOS活性增加。2)钙超载:生理状态下,胞浆内钙浓度约为10-7mol/L,而细胞外及胞浆内的钙储存系统(如内质网和线粒体)中钙浓度为10-3mol/L。但是再灌注后,钙离子向线粒体转移,导致线粒体功能障碍;钙离子浓度升高,可激活多种酶(如激活膜磷脂酶A2),同时促使心肌纤维过度收缩;通过Na+
/Ca2
+交换形成一过性内向电流,在心肌动作电位后形成延迟后除极,这是引起心律失常的原因之一。另外,它还促进ATP分解,使能量急剧减少等。3)微血管损伤和白细胞激活:正常情况下,中性粒细胞与血管内皮细胞的相互作用受内皮细胞上的阴性糖苷及内皮细胞产生的众多抗炎因子的抑制,中性粒细胞处于静止状态。缺血时,激活的中性粒细胞与血管内皮细胞发生固定粘附,导致微血管机械阻塞,并可释放出大量的炎性介质,不但可改变自身的结构和功能,而且使周围组织细胞受到损伤,发生无复流现象,致使细胞发生不可逆性损伤和坏死,即再灌流性心肌损伤。
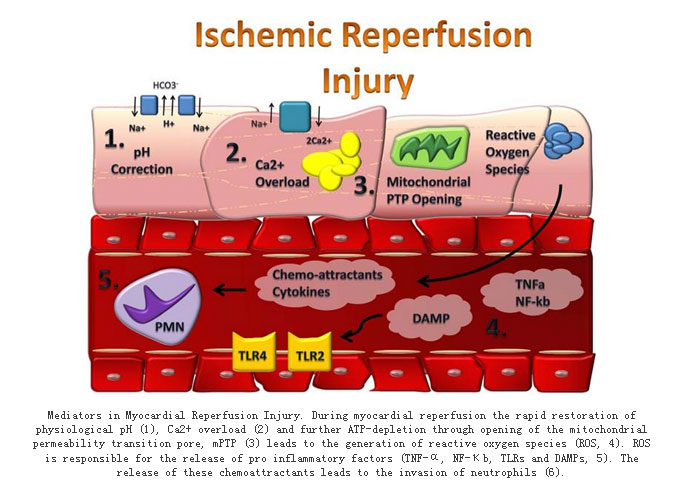
心肌缺血再灌注损伤的防治包括:清除自由基,“缺血后适应”处理,抑制Na+/H+ 、Na+
/Ca2+离子间的交换,应用腺苷受体激动剂、镁、他汀类、血管紧张素受体拮抗剂等药物。1)抗氧化治疗:自由基在许多疾病的发生、发展过程中扮演着重要的角色,因此自由基清除剂对疾病的控制显得尤为重要。然而,尽管有大量关于抗氧化剂的动物实验研究,对于临床上缺血再灌注损伤性疾病却没有常规应用,这并不是因为缺乏临床研究。离体鼠心脏缺血再灌注实验证明,同时应用超氧化物歧化酶、过氧化氢酶能明显减少心肌细胞凋亡,尽管理论与科学实验证明自由基清除剂或抗氧化剂的联合应用有效,但迄今为止,这些药物的联合应用在临床上尚未广泛开展,有待进一步的研究和验证。2)减轻钙超载:许多研究表明运用钠氢交换阻断剂和钠钙交换阻断剂进行药物预适应可以影响心肌缺血再灌注损伤的后果。3)缺血预处理(IPC)和药物预处理(PPC):心肌预处理是一种普遍和有力的抗缺血再灌注损伤的内源性保护机制,适当的药物预处理可有效调动该保护机制。近年来无数的动物模型均证实IPC能减小心肌梗死的范围,增加心肌收缩力及防止再灌注心律失常等。药物预处理(PPC)是根据IPC机制,通过药物激发或模拟机体内源性物质而呈现减弱MIRI的方法。其研究方法也是通过模拟IPC建立起来的。
研究模型
研究心肌缺氧复氧损伤及药物的干预作用,动物模型是不可缺少的。目前常用的造成心肌缺血再灌注整体动物模型的方法主要是冠状动脉结扎法。但由于动物个体的差异性、手术操作的不稳定性等因素造成了这种方法所制模型缺乏一定的稳定性,直接影响对药物干预作用的观察。体外培养心肌细胞,选择搏动规律、生长良好的细胞进行实验,造成心肌细胞缺氧/复氧病理模型,不仅避免了体内神经、体液的影响,而且也避免了整体动物模型的不稳定性,加强了模型的均一性,是筛选防止缺血性心脏病有效药物的良好体外病理模型。
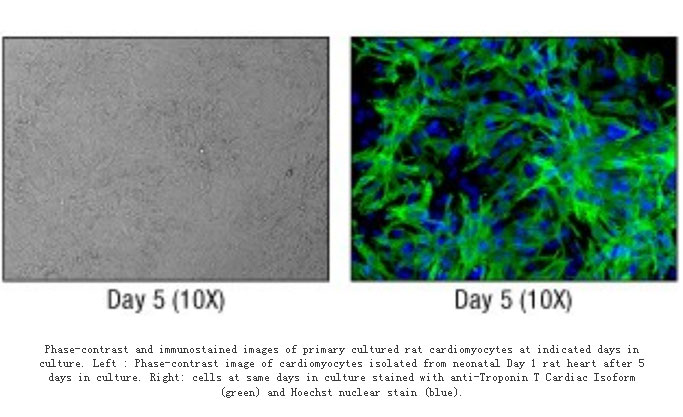
我们已经掌握了原代心肌细胞分离及培养的技术:取出生3天以内的SD大鼠乳鼠,酒精消毒,用眼科剪在胸骨角上2肋剪开胸腔,轻挤胸腔,挤出心脏,用镊子取心脏,将取下的心脏剪碎,用胰酶少量多次消化组织块,采用物理和化学方法去除成纤维细胞,纯化心肌细胞;用含15%FBS的DMEM培养基接种,在二氧化碳培养箱中培养。相差显微镜下观察刚提取的心肌细胞为球形、透亮、有立体感。12小时之后细胞基本贴壁,细胞逐渐伸出伪足,细胞之间逐渐建立联系;48小时细胞逐渐出现跳动。利用免疫荧光的方法检测提取细胞中cTNT表达。原代心肌细胞的观察如下图所示。
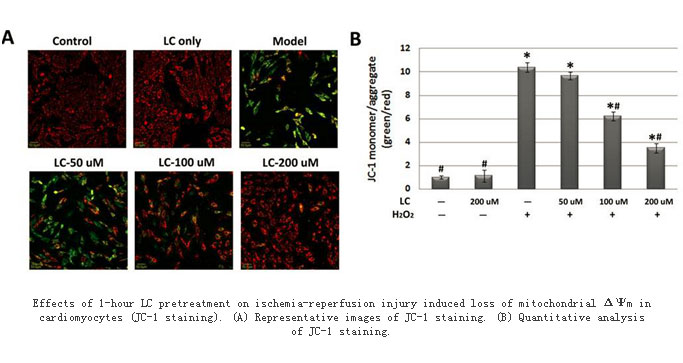
我们已经建立了心肌细胞缺氧复氧损伤模型,根据造成缺氧的原理可分为物理方法和化学方法。物理性缺氧法,即是将培养的心肌细胞,洗净旧培养基,给予预先排除氧气的无糖培养基,置密闭盒中持续通以95%N2+5%CO2混合气,通气15分钟后,密闭缺氧盒;4小时后打开缺氧盒,取出培养板,换为DMEM培养基继续培养。化学性缺氧法,即是在培养基质中加入化学物质,导致培养细胞缺氧的方法。常用的化学物质主要有氯化钴和连二亚硫酸钠。同时,我们选择合适的检测指标来评估和调整相关参数(如细胞密度、药物浓度、药物处理时间等)。根据目的不同,所选择的指标也不同,如反映细胞状态可直接在显微镜下观察,反映细胞活力可以用MTT法直接检测,反映细胞破损可用试剂盒检测细胞上清中的LDH或细胞内的MDA,反映细胞凋亡可用荧光染料检测。下图显示的是,利用JC-1染料评价心肌细胞线粒体膜电势在缺氧复氧损伤时的变化(LC代表某种药物)。
研究案例
目的:阐明PJ-34对心肌缺血再灌注损伤的保护作用
结论:PJ-34后处理显著改善了心肌的缺血再灌注损伤;PJ-34通过抑制AMPK激活而减轻细胞的氧化应激损伤
路线:
-
应用Langendorff离体心脏灌流系统构建大鼠全心缺血再灌注模型,并给以PJ-34后处理,观察其对缺血再灌注损伤的心肌是否具有保护作用
-
以外源性H2O2短时间刺激心肌细胞来构建心肌细胞的急性氧化应激损伤模型。研究PJ-34预处理对于H2O2诱导的心肌细胞损伤的保护作用,包括:提高细胞活性,改善线粒体功能,抑制凋亡相关蛋白激活
-
研究PARP-1/AMPK通路在H2O2诱导的心肌细胞氧化应激损伤中的作用。PARP-1和AMPK的抑制都可以减轻损伤,而AMPK激活剂AICAR则可以部分逆转PJ-34的有益作用
-
探讨细胞内PARP-1和AMPK两者之间的相互关系。外源性的活性蛋白PARP-1被转染进入细胞后,细胞内PARP-1和AMPK两种蛋白的表达和活性都增高,而免疫共沉淀实验也证明AMPK的激活与PARP-1的表达和活性直接正相关
核心文献
-
Hypoxia-Induced Signaling in the Cardiovascular System,Annu Rev Physiol.
2008; 70: 51–71.,http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2871679/
-
Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J
Clin Invest. 2013;123(1):92–100. http://www.jci.org/articles/view/62874
-
Myocardial reperfusion injury. N Engl J Med. 2007;357(11):1121–1135.
http://www.ncbi.nlm.nih.gov/pubmed/17855673
-
Impact of myocardial haemorrhage on left ventricular function and
remodelling in patients with reperfused acute myocardial infarction. Eur Heart
J. 2009;30(12):1440–1449. http://www.ncbi.nlm.nih.gov/pubmed/19346229
-
Effect of remote ischaemic preconditioning on myocardial injury in patients
undergoing coronary artery bypass graft surgery: a randomised controlled trial.
Lancet. 2007;370(9587):575–579. http://www.ncbi.nlm.nih.gov/pubmed/17707752